看点
这些数据为第一次对非洲猪瘟病毒进行比较基因组分析,第一个直接从自然感染的猪身上获得的未经细胞培养繁殖的非洲猪瘟病毒基因组序列。
这些数据还为研究非洲猪瘟病毒野生型基因组微进化提供依据,并为非洲猪瘟暴发早期的遗传变异研究提供了基础数据,为今后的基因组水平比较研究和流行病学调查提供了第一手资料。
在G2007/1基因组代表祖先序列的情况下, G-2008/1和G-2008/2基因组中的串联重复区A/T变异的出现和维持表明非洲猪瘟病毒对本地宿主群体可以快速稳定适应。
非洲猪瘟病毒基因组在位于左右可变区域的多基因家族(MGF)基因中表现出高度的多样性。
毒株比较
本文中两个非洲猪瘟病毒分离株(G-2008/1和G-2008/2)来自2008年格鲁吉亚初期爆发非洲猪瘟采集的PCR阳性猪血液。
/1和/2毒株的基因组序列已提交至GenBank数据库,接收号分别为MH910495和MH910496。
本文中的G-2008/1和G-2008/2的基因组序列与之前已公布的当前流行的非洲猪瘟病毒基因组序列(G-2007/1)进行比较。
分离株是从受感染的猪器官组织中获得、培养,并从原代猪骨髓细胞中获得的。
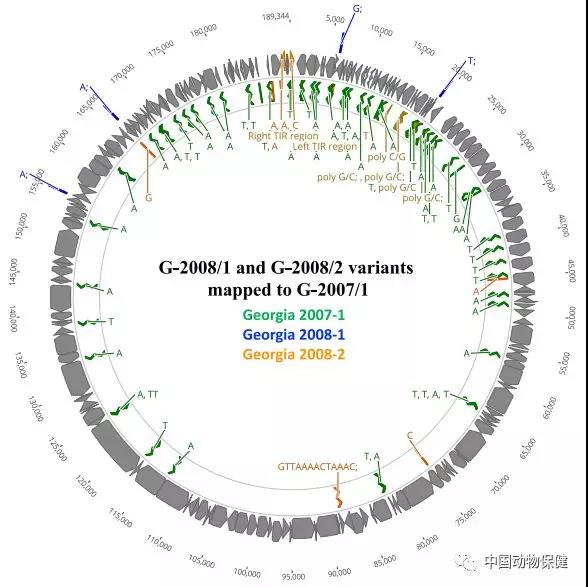
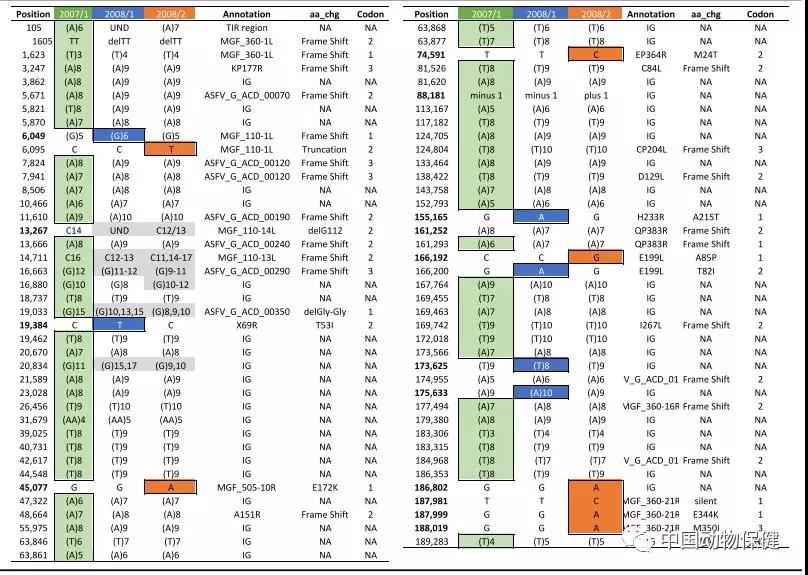
非洲猪瘟病毒基因组的结构和遗传多态性如图1和表1所示。这两个基因组与G2007/1参考基因组具有完全的共线性和相同的基因数量。
图1 G-2008/1、G-2008/2毒株与G-2007/1参考基因组的基因组多态性。
三个基因组中每一个差异都用不同的颜色来表示(G-2007/1=绿色,G-2008/1=蓝色,G-2008/2=橙色)
表1 非洲猪瘟病毒分离株2007/1、2008/1和2008/2之间的全基因组遗传多态性
表头分别是:
位点 2007/1 2008/1 2008/2 注释 突变类型 密码子 位点 2007/1 2008/1 2008/2 注释 突变类型 密码子
和G-2008/2基因组通过编码7个基因的变异来区分,7个基因包括MGF 110-1 L、X69R、MGF 505-10R、EP364R、H233R、E199L和MGF 360-21R,此外它们在左(n=6)和右(n=2)末端区域的8个串联重复区也有差异(表1)。
在传统的非洲猪瘟病毒亚型分析中使用的p72 (B646L)、P54 (E183L)、CVR (pB602L)和I73R/I329L串联重复区的分子靶点没有发生序列变异。
比较基因组分析还揭示了G2007/1参考基因组特有的56种独特的串联重复A/T-区变异(表1)。
图2
a,G-2008/2毒株的基因C315R(TFIIB样蛋白)和C147L(RNAPOL亚基6)之间的基因间区中C315R/C147L位点的串联重复插入的序列比对图。
b, 2007/1、2008/1和2008/2毒株的重复单元。
c,非洲猪瘟病毒各毒株中C315R/C147L位点的核酸序列比对。
b和c中标颜色的碱基为差异SNP位点
在G-2008/2基因组的基因C315R(TFIIB样蛋白)和C147L(RNApol亚基6)之间观察到一种新的基因间序列插入(GTTAAAACTAAAC)(图2a、b和c)。
短串联重复序列(图2b)构成较大间接重复结构的一部分(图2b)。该位点显示出整个非洲猪瘟病毒毒株拷贝单元的差异(图2c)。
整个毒株基因区的长度从20 bp(Ken05-Tk1)到93 bp(OURT和NHV)(图2b)。插入缺失界限延伸至C147L基因的末端。
串联重复区变异
格鲁吉亚非洲猪瘟病毒基因组中的替代突变和串联重复区变异发生在基因组的末端区域,特别是在LVR区中较高(图1)。
在G2007/1基因组代表祖先序列的情况下, G-2008/1和G-2008/2基因组中的串联重复区A/T变异的出现和维持表明非洲猪瘟病毒对本地宿主群体可快速稳定适应。
在G-2007/1基因组和G-2008/1和G-2008/2之间明显观察到串联重复区A/T变异,尽管在G-2007/1和本文报道的野生型G-2008/1和G-2008/2基因组进化的背景可能不同。
我们发现,在我们的分析中,选择2007/1基因组特有的串联重复区A/T位点也显示了先前研究中的变异,如末端多聚T、CP204L基因和负责在L60、E75和NHV28中融合MGF 110-13 L同系物的串联重复区。
此前,14种非洲猪瘟病毒蛋白(包括P54和E183L)中也描述了串联重复区的变异性。
在其他毒株中也发现在串联重复区存在显著的序列长度变异,如在L60和E7528毒株的M1249L基因中,在G-2007/1、G-2008/1和G-2008/2三个非洲猪瘟病毒基因组中表现出单等位基因多态。
分子标记的稳定性
我们发现以前的数据表明,体外繁殖可能改变目前用于非洲猪瘟病毒亚型分析的位点。我们对先前一项研究(描述了毒株BA71对Vero细胞的适应性)中产生的基因组序列数据进行分析,结果表明,适应性毒株BA71V特异地拥有一个新的CVR(PB602L)等位基因,该等位基因与葡萄牙毒株Por 63的等位基因相同。
在猪巨噬细胞和Vero细胞的复制过程中,在所选毒株的CVR (B602L)标记处也发现了同义突变。
除了在非洲猪瘟病毒亚型中的应用外,串联重复标记也用于分析其他双链DNA病毒的分子变异,包括丙型肝炎病毒、腺病毒、人类巨细胞病毒和疱疹病毒。该方法是一种可行的检测现有非洲猪瘟病毒标记稳定性的策略,也是与当前流行性非洲猪瘟病毒分离株追踪的未来分子标记。
对来自当前流行的一组地理上不同的纵向分离株进行全基因组变异分析将有助于识别新的更具流行病学信息的标记位点。这里报告的基因组(除了G-2007/1基因组外),为非洲猪瘟病毒亚型分类等工作提供了早期的基础数据。
在我们的研究中,跨越6个高变G/C区基因座的基因组区域的遗传结构变异已在之前报道过。重复序列位于串联A+T富集区的核心区,在MGF 110和MGF 300基因附近,这些基因共同形成LVR(38)中不同的大片段重复序列。
和G-2008/2基因组中所有剩余的串联G/C-区都含有少于10个残基,并且没有在样本中出现拷贝数变化。相比之下,基因组中的串联A/T区基因座没有检测到变异。与G-2007/1基因组相比,和G-2008/2基因组仅表现出两个不同的串联A/T区,并且各样本中未出现序列异质性。在两个分离株中,仅在基因组这一区域的串联区发生变异的机制尚不清楚。
野生型基因组同质性
由于DNA病毒拥有编码高保真DNA校对的修复酶,它们通常表现出较低的突变率。但是,非洲猪瘟病毒基因组独特地编码已知唯一的X型聚合酶(X-Pol)和一个DNA连接酶,这几个酶每个都表现出低保真度。目前已知的非洲猪瘟病毒DNA连接酶的体外保真度最低。非洲猪瘟病毒毒株显示的极端抗原多样性的遗传和表型基础尚未确定。
据推测,野外分离株中抗原多样性的提高是由X- Pol /DNA连接酶的活性引起的。先前的研究者假设Pol x/连接酶系统在碱基切除修复途径中起到突变DNA的作用,碱基切除修复途径中由于DNA修复错误的增加而发生快速遗传漂变。
关于Pol x保真度也有不同的报道。据推测,这些不一致的报道是由于已发表的研究之间的酶氧化还原状态差异造成的,还原形式的酶显示出更高的保真度。
我们推测,掌握和了解非洲猪瘟病毒DNA突变模型可发现更大的样本内全基因组变异和提高野生型分离株之间的基因组多态性。
我们的数据表明宿主内和基因组间序列的同质性。这些数据与近期其他研究中描述的非洲猪瘟病毒基因组复制中DNA保真度的因素一致,包括病毒II类无嘌呤/无嘧啶内切酶(E296R蛋白)的活性和最近报道的POL X在体内的修复作用。
讨论和结论
本文报道的基因组可为非洲猪瘟病毒分离株的流行病学追踪提供参考信息,并可为急性野生型感染期间的病毒微进化研究提供思路。
位点的新等位基因目前是当前流行谱系分离出的最早观察到的遗传变异。
非洲猪瘟病毒基因组在位于左右可变区域的多基因家族(MGF)基因中表现出高度的多样性。
在区分G-2008/1、G-2008/2毒株基因组的15个SNP中,有11个是非同义的。其余4个单核苷酸多态性代表了基因间区串联重复区的变异。我们还可以进一步拓展分析基因组数据的同义变异。
我们的结果证实了深度测序作为一种高度信息化的无培养方法的直接从PCR阳性血液中进行非洲猪瘟病毒基因组研究的实用性。来自这一流行病的更多基因组数据将更好地促进有针对性的病毒分析工作的开展,并在这一流行病持续过程中提供更强有力的流行病学追踪手段。